See the5-yeardata
After just one treatment, AMTAGVI® delivered
deep and durable responses1
Studied in patients with previously treated unresectable or metastatic melanoma
AMTAGVI was evaluated in Study C-144-01, a global, multicenter, multicohort, open-label, single-arm clinical trial. Cohort 4 of Study C-144-01 enrolled patients with advanced melanoma who have previously been treated with at least 1 systemic therapy, including a PD-1 blocking antibody, and if BRAF V600 mutation-positive, a BRAF inhibitor or BRAF inhibitor with MEK inhibitor. The primary endpoint was IRC-assessed ORR by RECIST v1.1. Select secondary endpoints included DOR and OS. Seventy-three patients received AMTAGVI, which was manufactured at an approved facility at the recommended dose.
Supportive Pooled Data (n=153) includes Cohort 4 plus Cohort 2 of Study C-144-01, which enrolled patients who met the same primary eligibility criteria.1,2
Results for AMTAGVI include long-term data
with 5 years of follow-up1,3
Cohort 4 Pivotal (N=73)
ORR 31.5%
(95% CI: 21.1, 43.4)
mDOR NR
(Range: 1.4+, 26.3+)
18.6 months
(potential)
Potential median study follow-up
*95% CI: 4.1, NR.1
18.6 months
(potential)
Supportive Pooled Analysis (n=153)
ORR 31.4%
(95% CI: 24.1, 39.4)
mDOR NR
(Range: 1.4+, 45.0+)
21.5 months
(potential)
Potential median study follow-up
21.5 months
(potential)
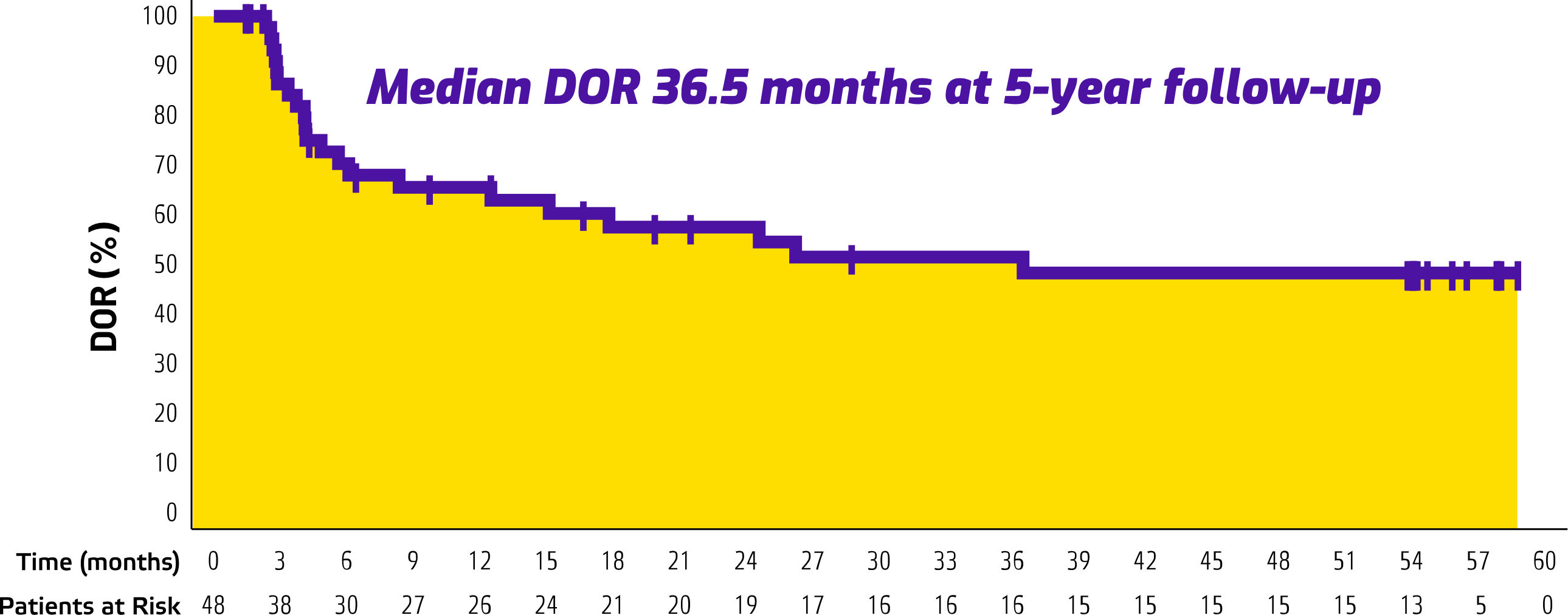
5-Year Follow-Up Data (n=153)
ORR 31.4%
(95% CI: 24.1, 39.4)
mDOR 36.5 months
(95% CI: 8.3, NR)
Median study follow-up: 57.8 months

*95% CI: 4.1, NR.1
Treatment-Related Mortality AMTAGVI is associated with treatment-related mortality. In the clinical trial, the treatment-related mortality rate was 7.5% (N=160), including
2 deaths during the lymphodepleting period, 6 deaths within 30 days, and 4 deaths 38 to 150 days following AMTAGVI administration. Adverse reactions associated with these deaths included severe infections (sepsis, pneumonia and encephalitis), internal organ hemorrhage (abdominal hemorrhage and intracranial hemorrhage), acute renal failure, acute respiratory failure, cardiac arrythmia, extensive ascites and liver injury and bone marrow failure. Because clinical trials are conducted under widely varying conditions, treatment-related mortality rates observed in the clinical trials of a drug may not reflect the rates observed in practice.1
CI, confidence interval; DOR, duration of response; IL-2, interleukin-2; IRC, independent review committee; mDOR, median duration of response; MEK, mitogen-activated protein kinase kinase; NR, not reached; ORR, objective response rate; OS, overall survival; PD-1, programmed death receptor-1; RECIST, Response Evaluation Criteria in Solid Tumors.